
Shashi B. Mishra
Assistant Research Scientist, Department of Electrical & Computer Engineering,
University of Maryland, College Park
About
I am a theoretical condensed matter physicist and computational materials scientist with expertise in first-principles simulations, many-body theory, and high-performance computing. My work focuses on understanding quantum materials through electron-phonon interactions, with particular interests in superconductivity beyond the Migdal approximation, transport in topological semimetals, and light-induced magnetic phenomena in metals. I am also a contributor to the open-source EPW code, where I have implemented electron-phonon vertex corrections and efficient two-level MPI parallelization strategies for superconductivity.
Featured research
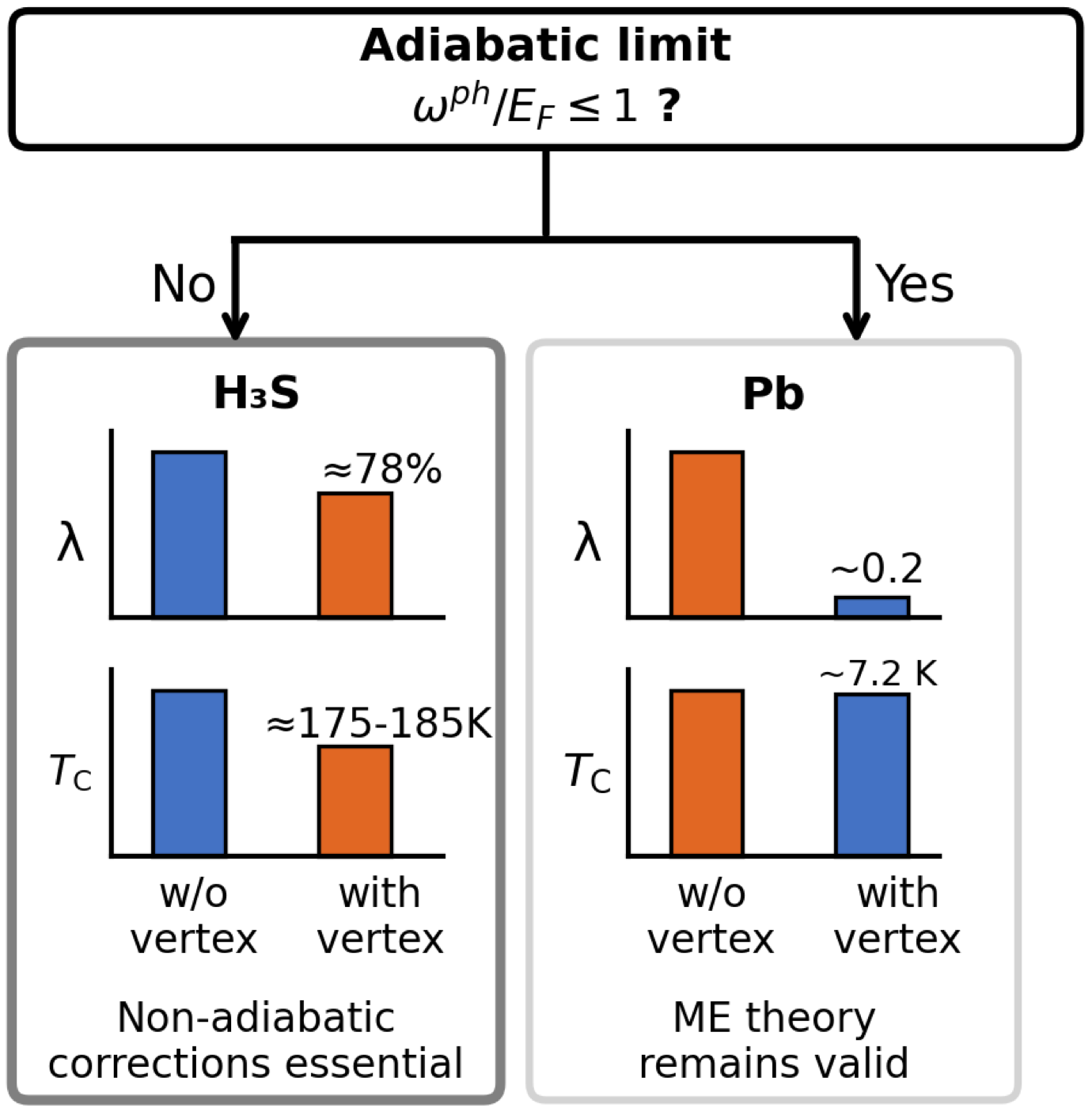
Vertex corrections in H3S
npj Comput. Mater. 11, 342 (2025)
First-principles electron-phonon vertex corrections reduce the predicted Tc of H3S by roughly a quarter, while leaving conventional superconductors like Pb essentially untouched — refining the Migdal framework for high-pressure hydrides.
Read the paper →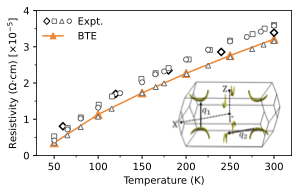
Transport in topological materials
Phys. Rev. B 113, 045202 (2026) · Phys. Rev. B 112, 104311 (2025)
Phonon-limited carrier transport in Weyl semimetals — established first for TaAs (2025) and then traced systematically across the TaAs family (2026) — resolving how Berry-curvature-rich bands and electron–phonon scattering jointly shape conductivity.
Read the paper →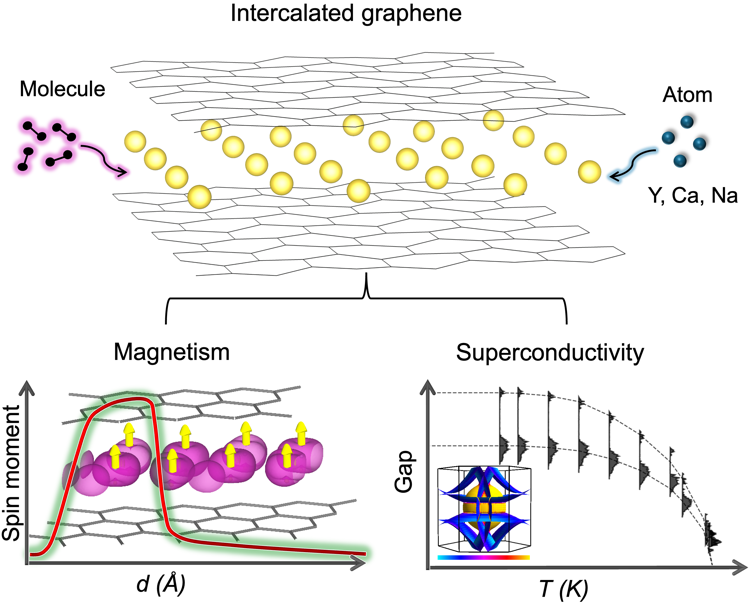
Na-intercalated graphite superconductors
Phys. Rev. B 110, 174508 (2024)
A stability–superconductivity map for compressed Na-intercalated graphite — charting the stoichiometries and pressures that maximize phonon-mediated Tc within the Migdal regime to guide experimental synthesis.
Read the paper →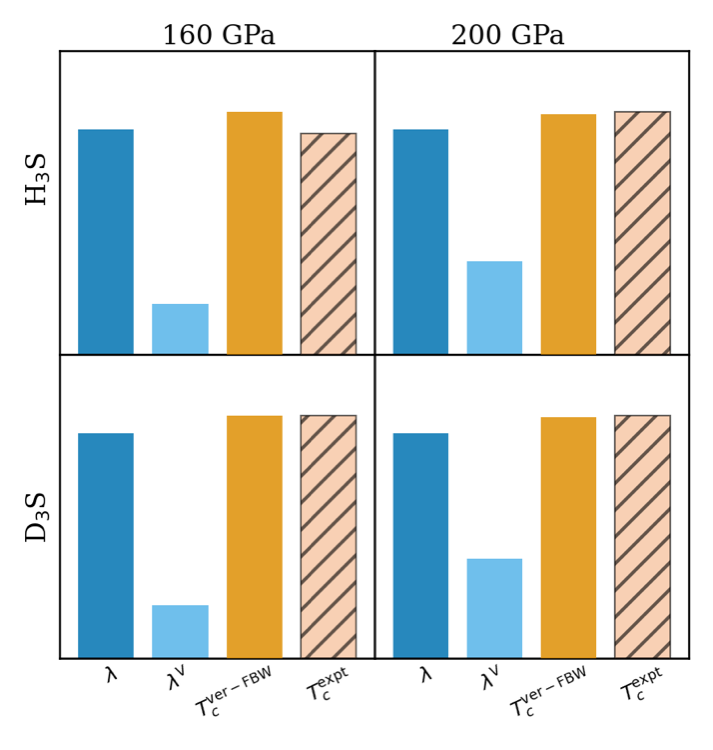
Non-adiabatic & anharmonic H3S / D3S
Annalen der Physik 538, e00553 (2026)
Full-bandwidth ab initio Eliashberg calculations that quantify how non-adiabatic electron-phonon coupling and phonon anharmonicity jointly reshape the isotope effect and Tc in H3S and D3S.
Read the paper →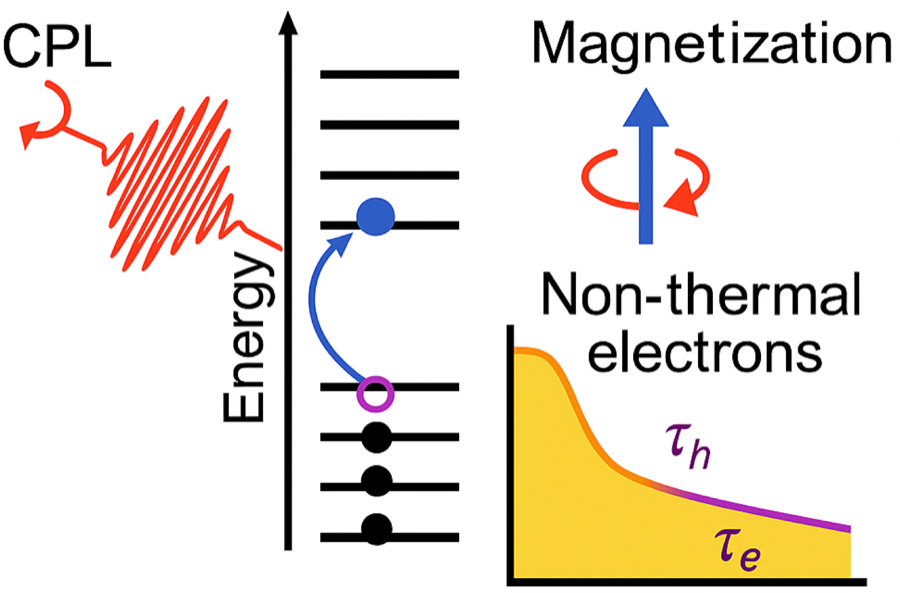
Inverse Faraday effect in metals
Phys. Rev. B 111, 174413 (2025) · Phys. Rev. B 107, 214432 (2023) · Phys. Rev. Mater. 7, 125202 (2023)
A gauge-invariant first-principles theory of light-induced magnetization — formulated for non-magnetic metals using degenerate perturbation theory with Wannier interpolation (2023), validated by transient-ellipsometry measurements (2023), and traced systematically across the 3d, 4d, and 5d series (2025) to enable ultrafast optical control of magnetism.
Read the paper →
Energy materials by design
ACS Appl. Energy Mater. 4, 7786 (2021)
First-principles design of two-dimensional anodes and cathodes — Si2BN, graphyne, graphdiyne — for next-generation Al- and Mg-ion batteries, balancing capacity, diffusivity, and structural stability.
Read the paper →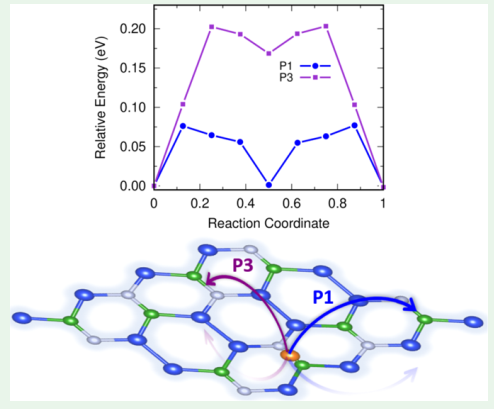
Si2BN for energy storage devices
ACS Appl. Nano Mater. 3, 9055 (2020) · J. Energy Storage 77, 109913 (2024)
The unique 2D Si2BN sheet is exploited as an anode for Mg-ion (2020) and high-capacity Al-ion (2024) batteries — its semimetallic electronic structure, low Mg/Al migration barrier, and high theoretical capacity enable fast charge/discharge cycles suitable for high-performance anodes.
Read the paper →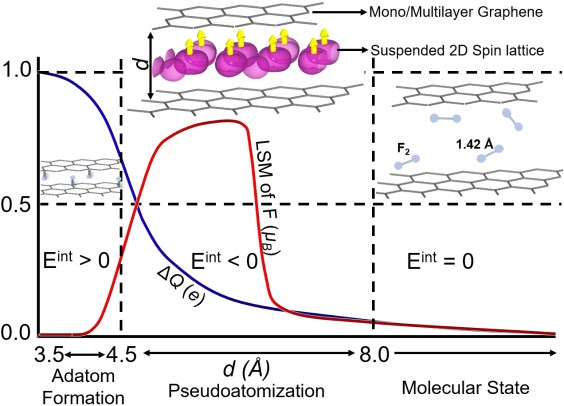
Suspended spin lattice in fluorine–graphene
Phys. Rev. Mater. 4, 074411 (2020)
Fluorine intercalated between graphene layers is pseudoatomized through bond stretching and charge transfer, stabilizing a spin-polarized ground state with long-range magnetic order — an artificial van der Waals spin lattice with the graphene host retaining its pristine bands.
Read the paper →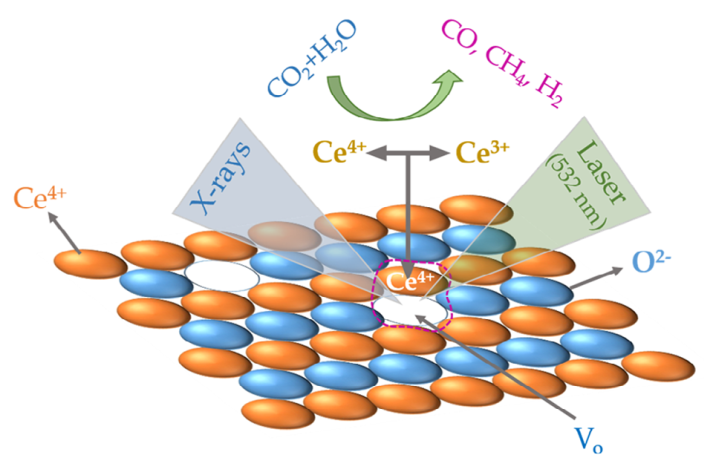
Catalytic activities of defect / heterostructure CeO2
Adv. Funct. Mater. e13933 (2025) · ACS Appl. Mater. Interfaces (2025)
First-principles analysis of photo-excited defect dynamics in CeO2 and of the electrostatic-self-assembled CeO2/g-C3N4 heterojunction — two routes that respectively enhance CO2 photoreduction and photocatalytic hydrogen evolution on ceria surfaces.
Read the paper →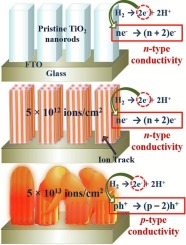
Irradiation-induced activity in TiO2
Mater. Sci. Eng. B 326, 119210 (2026) · Nanoscale Adv. 4, 241 (2022)
Swift heavy-ion irradiation tailors the crystallinity, morphology, and electronic structure of TiO2 nanorods via a thermal-spike mechanism — unlocking anomalous gas-sensing characteristics and modified catalytic activity beyond what pristine growth can deliver.
Read the paper →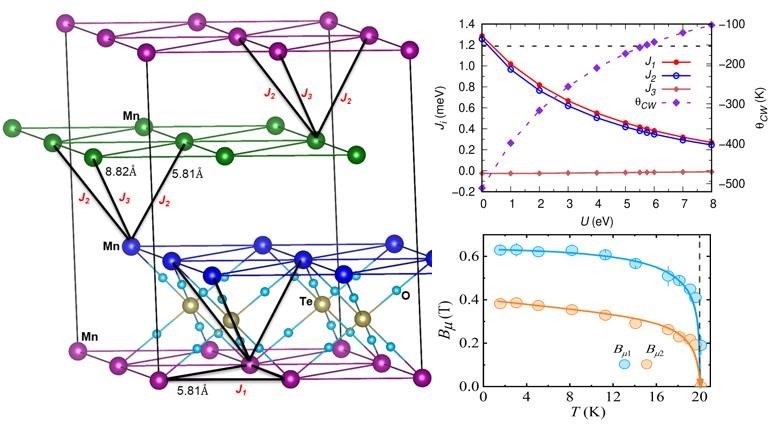
Magnetic order in Ba2MnTeO6
Sci. Rep. 11, 6959 (2021)
Experiment and DFT+U together resolve competing intra- and inter-layer AFM exchanges on the Mn2+ triangular lattice of Ba2MnTeO6, stabilizing a 3D long-range magnetic ordering that matches the measured Curie–Weiss temperature at U = 5.75 eV.
Read the paper →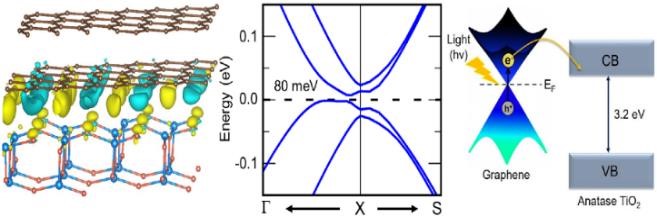
Graphene / TiO2 interface
Appl. Surf. Sci. 542, 148709 (2021)
A low-strain commensurate graphene/TiO2 interface preserves graphene's gapless Dirac bands under strain; in the AB-stacked bilayer, interfacial charge transfer opens a narrow bandgap — enabling visible-light-sensitized photocatalysis.
Read the paper →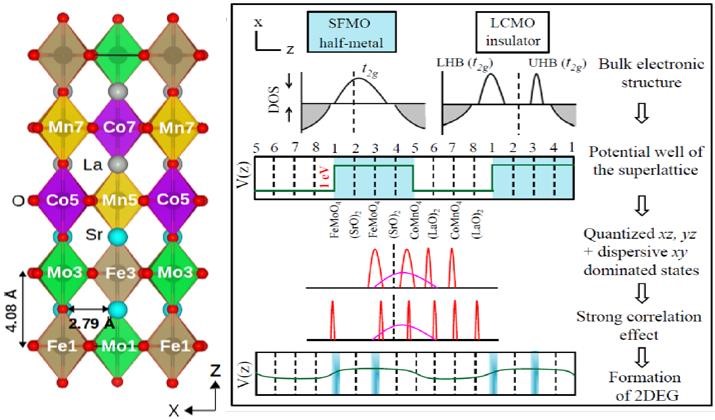
Spin-polarized 2DEG in oxide heterostructures
Phys. Rev. B 98, 115155 (2018)
A non-polar oxide heterostructure Sr2FeMoO6/La2CoMnO6 realizes a spin-polarized two-dimensional electron gas through quantum confinement and orbital manipulation — a route distinct from LaAlO3/SrTiO3-like polar interfaces.
Read the paper →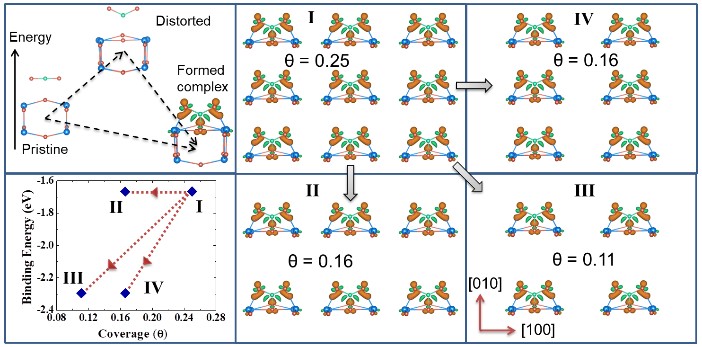
Three-state model of CO2 on TiO2(001)
Phys. Rev. Mater. 2, 115801 (2018)
A three-state model couples surface deformation, charge transfer, and molecular-orbital bonding to explain CO2 adsorption on anatase TiO2(001) — predicting uniaxial, long-ranged interactions and strong binding-energy anisotropy confirmed by density-of-states analysis.
Read the paper →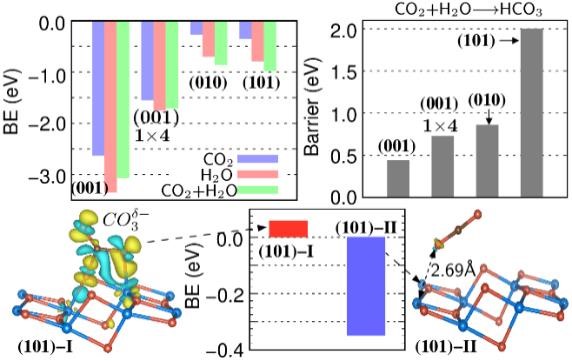
Facet-dependent CO2 catalysis on TiO2
Appl. Surf. Sci. 531, 147330 (2020)
Ranking CO2, H2O, and co-adsorption-driven bicarbonate formation across low-index anatase TiO2 facets — including the reconstructed (1×4)-(001) — with the change in exchange–correlation energy serving as a quantitative bond-strength indicator.
Read the paper →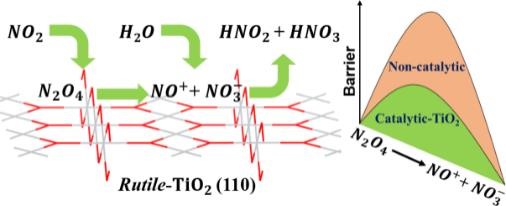
NO2 dissociation on rutile TiO2(110)
J. Phys. Chem. C 124, 8786 (2020)
DFT reaction-pathway analysis for NO2 on rutile TiO2(110): NO2 dimerizes into a short-lived N2O4 that dissociates to ionic species and, with H2O present, converts to HONO and HNO3 — a roadmap for toxic-gas conversion on oxide surfaces.
Read the paper →Research themes
Superconductivity & electron-phonon coupling
Phonon-mediated pairing beyond Migdal, vertex corrections, anharmonicity, and first-principles design of ambient-pressure superconductors.
Topological transport & Berry curvature
Phonon-limited carrier transport in Weyl and Dirac semimetals; Berry-curvature-driven anomalous responses from first principles.
Light–matter interaction
Inverse Faraday effect across 3d–5d metals, non-thermal phase transitions, and ultrafast optical control of magnetism.
Energy materials & catalysis
Design of two-dimensional battery anodes (Si2BN, graphdiyne, graphyne) and catalytically active oxide surfaces and interfaces.
Selected publications
- Comparative study of phonon-limited carrier transport in the Weyl semimetal TaAs family. Phys. Rev. B 113, 045202 (2026).
- Nonadiabatic and anharmonic effects in high-pressure H3S and D3S superconductors. Annalen der Physik 538, e00553 (2026).
- Electron-phonon vertex correction effect in superconducting H3S. npj Computational Materials 11, 342 (2025).
- Inverse Faraday effect in 3d, 4d, and 5d transition metals. Phys. Rev. B 111, 174413 (2025).
- Phonon-limited carrier transport in the Weyl semimetal TaAs. Phys. Rev. B 112, 104311 (2025).
- Cubic BeB2: a metastable p-type conductive material from first principles. Phys. Rev. B 112, 155206 (2025).
- Stability–superconductivity map for compressed Na-intercalated graphite. Phys. Rev. B 110, 174508 (2024).
- Spin contribution to the inverse Faraday effect of nonmagnetic metals. Phys. Rev. B 107, 214432 (2023).
- Graphdiyne — a promising cathode material for aluminium dual-ion batteries. ACS Appl. Energy Mater. 4, 7786 (2021).
- Si2BN nanosheets as anode materials for magnesium-ion batteries. ACS Appl. Nano Mater. 3, 9055 (2020).
See the full publication list for all work.
Curriculum vitae
Contact
Department of Electrical & Computer Engineering
University of Maryland, College Park, MD 20742